最近Orthofinder2开始陆续更新,文章已经投发到biorkix上,在网上搜了一圈,关于Orthofinder的使用的中文教程几乎是空白的,这周就借此机会和大家一起来学习一下这款软件。
Orthofinder介绍
OrthoFinder是比较基因组学中的实用的,运行快速,准确的全面的工具。它的主要功能是,找到了正交群和直系同源物,推断出所有正交群的根基因树,并识别那些基因树中的所有基因重复事件。它还为所分析的物种推断出有根的物种树,并将基因重复事件从基因树比对到物种树的分支中。
另外,OrthoFinder还为比较基因组分析提供全面的统计数据。相比OrthoMCL, OrthoFinder使用简单安装都很简单,安装只需要用过conda就行,运行只需一组FASTA格式的蛋白质序列文件(每个物种一个)。
基础知识介绍
为了让大家更好的理解使用Orthofinder,在正式介绍其使用的流程前,先来回顾一下与之相关的生物学知识。
直系同源物和旁系同源物
Orthologs(直系同源物)是在两个物种的最后共同祖先(LCA)中来自单个基因的一对基因。直系同源物是同源性基因,是物种形成事件的结果。Paralogs(旁系同源物)是同源基因,是重复事件的结果。下图就可以看到,不同物种间的alpha-chain gene互为Orthologs(直系同源物)。这时候可以引用一个新名词orthogroup (正交群)就用来形容自一组物种的LCA中的单个基因的基因组(在图中就是alpha chain gene)。然后同一物种间alpha 和beta chain gene互为Paralogs(旁系同源物)。最后所有这些关系都可以由OrthoFinder来识别。
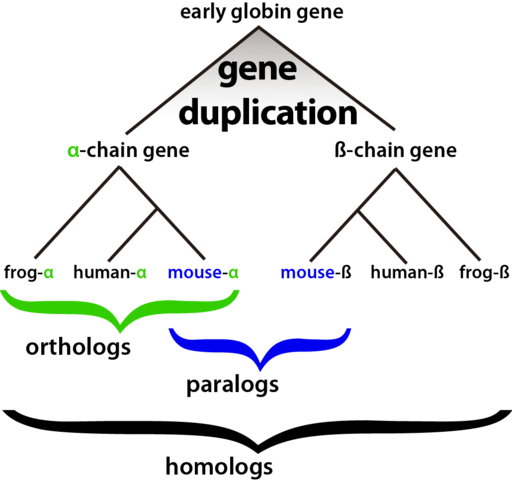
为什么要研究正交群?
- 正交群中的所有基因都来自单个祖先基因。因此,正交群中的所有基因都有类似的序列和功能。由于基因重复和丢失在进化中经常发生,一对一的直系同源物很少见,通过分析orhtogroup所有直系同源的情况(一对一,多对一,多对多),我们可以分析数据的所有情况。
- 直系同源物是由系统发育定义的。它不能通过氨基酸含量,密码子偏好,GC含量或其他序列相似性测量定义。在没有系统发育的情况下使用这些分数来定义,直向同源物的方法只能提供猜测。从序列相似性提供粗略的类比猜测直系同源,类似于从气味中猜测颜色。因此,真正识别直向同源物的唯一方法是通过分析系统发育树。确保直系分配正确的唯一方法是对所考虑的物种的最后共同祖先的单个基因下降的所有基因进行系统发育重建。这组基因是正交群。因此,定义直系同源的唯一方法是分析正交群。
安装
废话说了一大篇,是时候开始安装软件了。在这里推荐大家使用conda来安装,简单明了,不用担心其他Dependencies的安装
conda install orthofinder
接着准备好测试数据,为了展示的方便,我将原始测试数据重新命名:
##重命名
cd /work1/ExampleData
mv Mycoplasma_agalactiae.faa Maga.faa
mv Mycoplasma_gallisepticum.faa Mgal.faa
mv Mycoplasma_genitalium.faa Mgen.faa
mv Mycoplasma_hyopneumoniae.faa Mhyo.faa
##创建新的数据目录
mkdir Mycoplasma/
cp *faa Mycoplasma/
正式运行Orthofinder,相当简单的操作,-f输入目录,里面包含你需要运行的蛋白质fasta文件, -t 所用到的CPU数目。基本的用法就如下,更多的可以去manual中查看。
orthofinder -f Mycoplasma/ -t 2
生成的结果会存储于Results_XXX文件中,现在简单看看里面有啥。
正交群以两种不同的格式返回,一种是制表符分隔的表格格式(* .csv),另一种格式与orthoMCL(* .txt)的输出格式相同。还应该有一个名为WorkingDirectory的目录,其中包含运算过程的中间文件,例如blast结果。
cd Mycoplasma/Results_Nov10/
ll -thrl
-rw-r----- 1 hhu hhu 12K Nov 10 16:50 Orthogroups.GeneCount.csv
-rw-r----- 1 hhu hhu 59K Nov 10 16:50 Orthogroups.csv
-rw-r----- 1 hhu hhu 89K Nov 10 16:50 Orthogroups.txt
-rw-r----- 1 hhu hhu 110 Nov 10 16:50 Orthogroups_SpeciesOverlaps.csv
-rw-r----- 1 hhu hhu 33K Nov 10 16:50 Orthogroups_UnassignedGenes.csv
drwxr-x--- 6 hhu hhu 4.0K Nov 10 16:50 Orthologues_Nov10
-rw-r----- 1 hhu hhu 2.5K Nov 10 16:50 SingleCopyOrthogroups.txt
-rw-r----- 1 hhu hhu 1.5K Nov 10 16:50 Statistics_Overall.csv
-rw-r----- 1 hhu hhu 2.5K Nov 10 16:50 Statistics_PerSpecies.csv
drwxr-x--- 2 hhu hhu 4.0K Nov 10 16:50 WorkingDirectory
使用R简单分析Orthofinder的结果
安装加载以下R包
install.packages("micropan")
install.packages("phangorn")
library(micropan)
library(phangorn)
读取Orthofinder中正交群的文件。结果文件将正交群存储为制表符分隔表,其中每一行是正交群,其名为OG,后跟7位数字(例如OG0000123)。表中的每列对应一个物种,并且与fasta文件具有相同的名称。表中的单元格包含序列ID。由于正交组可以包含来自单个物种的几个基因(即,旁系同源物),因此表中的单个cell可以包含由逗号分隔的几个序列ID。下面是读取结果文件的例子:
# Define orthofinder results directory (may vary)
resultsDir <- "orthofinder/Mycoplasma/Results_Oct22"
grpsTbl <- read.table( file.path(resultsDir,"OrthologousGroups.csv"),
header=T, sep = "\t", row.names = 1, stringsAsFactors=F)
# Split the comma-separated paralogs
grps <- strsplit( as.matrix(grpsTbl),", ")
# grps is now a list of character vectors..
# Add dimensions to grps:
dim(grps) <- dim(grpsTbl)
# Add row and column names:
dimnames(grps) <- dimnames(grpsTbl)
获取特定正交群的信息:
grps["OG0000018", ]```
可以清晰的看到,在对应的fa文件中,在该特定的正交群OG000018下所包含的fasta序列,其结果如下:
## $Maga.faa
## character(0)
##
## $Mgal.faa
## [1] "Mgal_AAP56913.2" "Mgal_AAP56907.2" "Mgal_AAP56908.2"
##
## $Mgen.faa
## [1] "Mgen_AAC71531.2" "Mgen_AAC71529.1" "Mgen_AAC71563.2"
##
## $Mhyo.faa
## character(0)
接着让我们来看开正交群的数目,为了获得特定正交群的序列,需要在每个正交群中构建每个物种的基因数量的矩阵:
grpSize <- apply(grps,2,sapply,length)
我们可以在正交群OG0000018中检查每个物种中的旁系同源物的数量:
grpSize["OG0000018", ]
## Maga.faa Mgal.faa Mgen.faa Mhyo.faa
## 0 3 3 0
我们还可以绘制每个正交群中存在的基因数量:
plot(table(apply(grpSize>0,1,sum)),ylab="no. orthogroups",xlab="no. genomes represented")
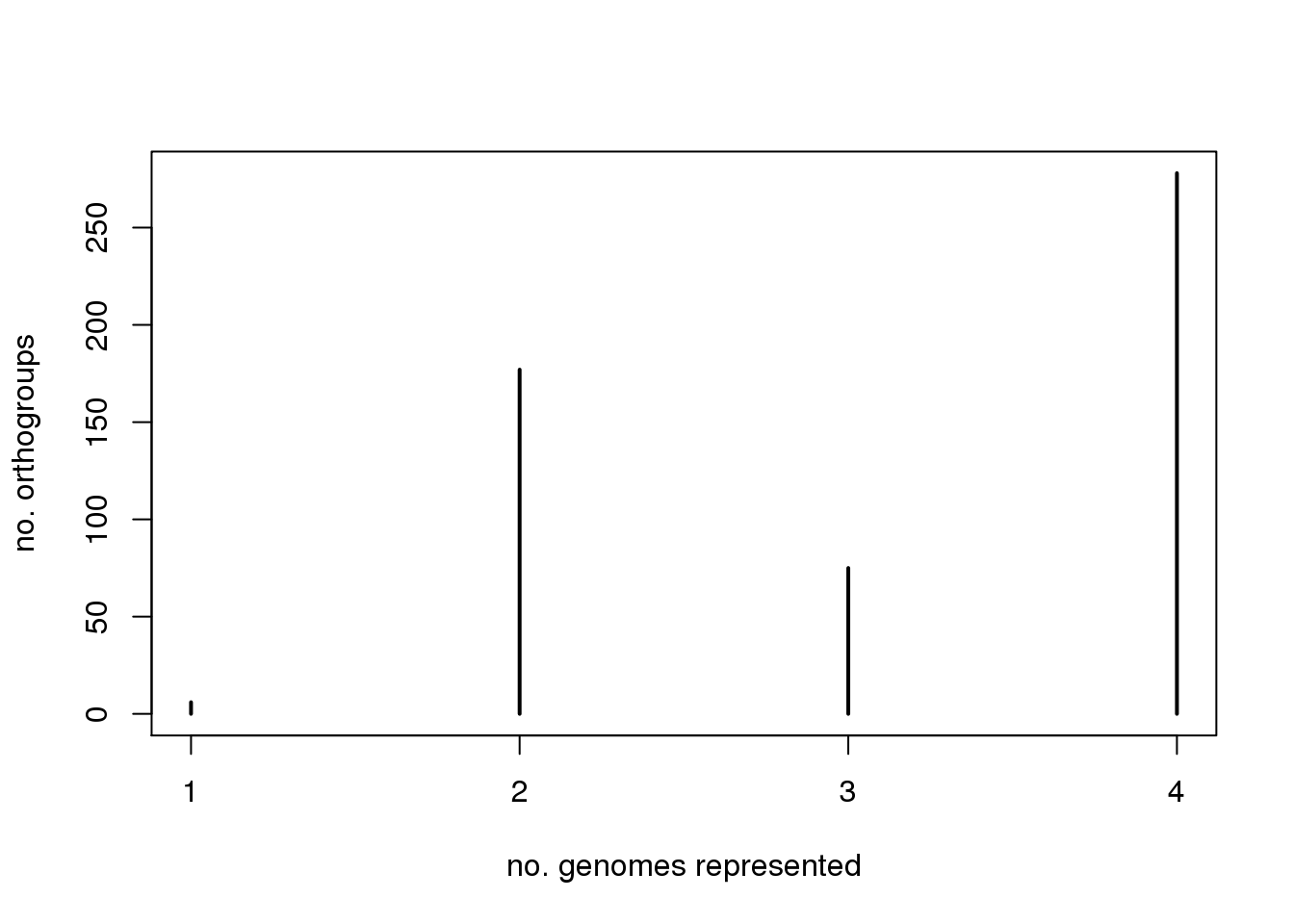
另外一写有趣的事我们可以查看,每个物种中只有一个直系同源的正交群(1:1:1:1正交群):
sum(apply(grpSize==1,1,all)) # number of 1:1:1:1 groups
## [1] 254
正好只存在于两个物种(1:1:0:0正交群)的正交群:
sum(apply(grpSize==1,1,sum)==2 & apply(grpSize > 1,1,sum)==0)
## [1] 155
让我们找出这个数字背后所对应的物种:
# get the 1:1:0:0 groups
is1100 <- apply(grpSize==1,1,sum)==2 & apply(grpSize > 1,1,sum)==0
spcs <- sub("\\.faa","",colnames(grps)) # get species names
# Count the combination of species-pairs in the 1:1:0:0 groups
table(apply(grpSize[is1100, ], 1, function(grpSizeRow){
paste(spcs[ grpSizeRow==1 ], collapse="&")
}))
##
## Maga&Mgal Maga&Mgen Maga&Mhyo Mgal&Mgen Mgal&Mhyo Mgen&Mhyo
## 15 2 57 70 8 3
基本的用法和分析就讲解到这里,当然这只是最基本的东西。其结果涉及到的下游分析还有很多很多,例如构建直系同源基因树等等的知识,大家可以自行进行探讨。
